
Navigating the Frontier of Proteomics: Intact Protein Mass Spectrometry vs. Native Protein Mass Spectrometry
Matthew Crawford, MS
Proteomics, the study of proteins and their functions within organisms, has revolutionized our understanding of biological systems. Mass spectrometry (MS) plays a pivotal role in proteomic research, enabling the identification, characterization, and quantification of proteins with unparalleled precision.
Within the realm of MS-based proteomics, two prominent techniques stand out: intact protein mass
spectrometry and native protein mass spectrometry. Let’s embark on a journey to unravel the intricacies that set these methodologies apart.
Intact Protein Mass Spectrometry
Intact protein mass spectrometry involves the direct analysis of proteins in their entirety, without nzymatic digestion or fragmentation. This approach provides valuable insights into protein size, structure, and post-translational modifications (PTMs).
In intact protein MS, denatured proteins are ionized and introduced into the mass spectrometer intact. The resulting mass spectra reveals peaks corresponding to the different charge states of the intact proteins, allowing for the determination of their molecular weights and heterogeneity. Intact protein MS is particularly useful for studying intact protein structure, determining protein stoichiometry in odification reactions (both synthetic and cell-based), and detecting PTMs on intact proteins.
Native Protein Mass Spectrometry
Native protein mass spectrometry, on the other hand, focuses on preserving the native structure and
non-covalent interactions of proteins during the ionization process. Unlike conventional intact MS methods that often denature proteins, native MS techniques aim to maintain the protein’s native, functional conformation and interactions with ligands, cofactors, or other proteins.
In native protein LC-MS, proteins are separated in the native state (usually by SEC in an MS compatible
buffer) and are then gently ionized under conditions that preserve their non-covalent interactions. This
allows for the detection of intact protein complexes, protein-ligand (& cofactor) interactions, and oligomeric states in their native physiological environment. Native MS is particularly valuable for studying
protein-protein interactions, protein folding dynamics, and the assembly of macromolecular complexes.
Key Differences and Applications
While both intact protein MS and native protein MS share the common goal of probing proteins at the
molecular level, they differ in their approach and applications:
Scope of Analysis: Intact protein MS primarily focuses on determining the molecular weight and heterogeneity of intact proteins, including the identification of PTMs and protein complexes. In contrast,
native protein MS emphasizes the preservation of native protein structure and interactions for studying
protein assemblies and interactions.
Sample Preparation: Intact protein MS often requires minimal sample preparation, making it suitable for high-throughput analysis of intact proteins. In contrast, native protein MS may involve more intricate sample preparation steps to maintain the native conformation of proteins and their complexes.
This also involves more extensive MS source optimization to ensure native state is maintained during ionization.
Structural Insights: Intact protein MS provides insights into protein size, heterogeneity, and PTMs, while native protein MS offers information about protein folding, oligomeric states, and non-covalent interactions.
To further illustrate the differences, let us look at Bovine Serum Albumin (BSA) and Equine Myoglobin as examples.
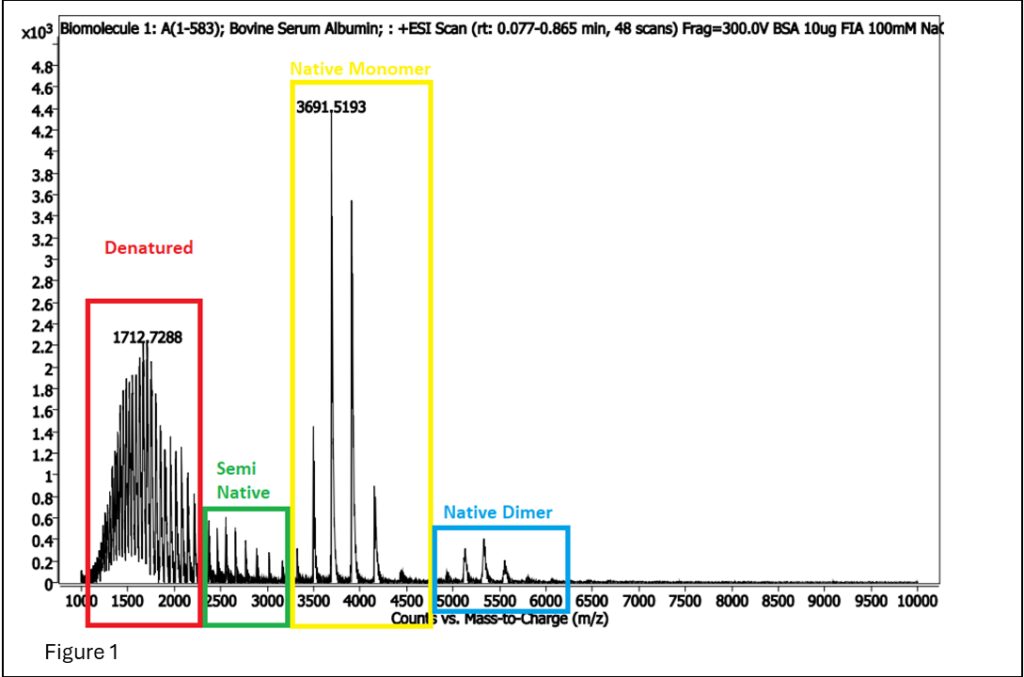
Figure 1: BSA spectrum showing areas that are denatured and native (generally referred to as intact protein analysis), and dimerized native.
In Figure 1, the signal for the denatured is cleaner and more robust than the signal for native MS, so why would we want to do native MS? The answer is… cofactors!! Cofactors are molecules that interact specifically with a protein, enabling its proper function. As such, native MS is often used to determine if a protein treatment or purification breaks the protein-cofactor association.
Myoglobin is a good example of how protein-cofactors relationships can require native MS analysis, as each myoglobin protein contains one Heme cofactor group for binding of oxygen molecules in muscle.
So, how can we tell if the myoglobin is in native state with the cofactor?
MS will tell us! Heme weighs 615 daltons, so the m/z +1 is 616, making it easily visible with an MS.
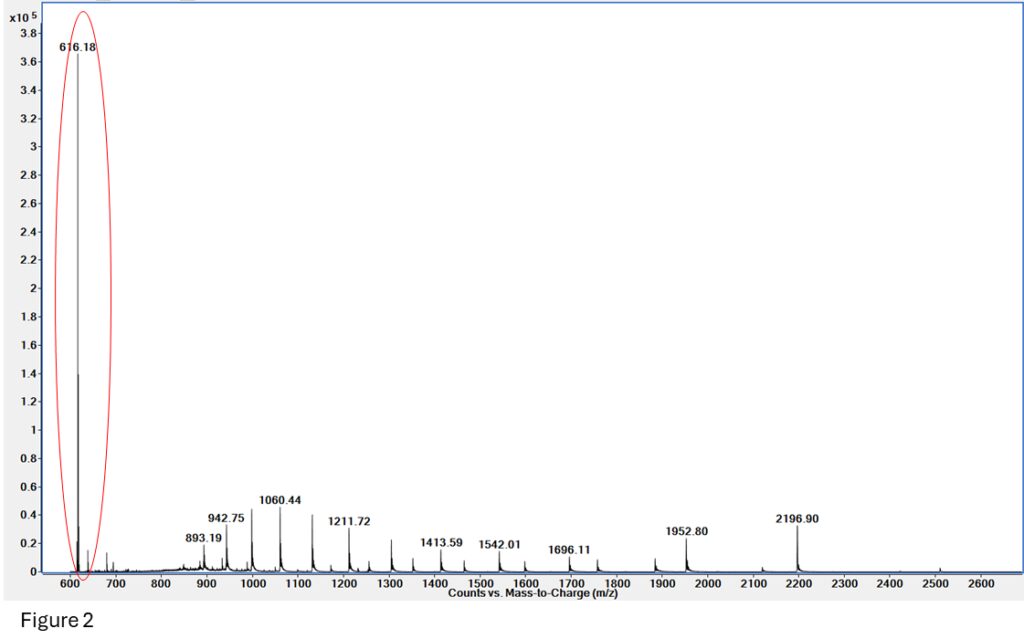
Figure 2: The Heme group at 616.18 is clearly visible on this spectrum, which also includes some native and denatured myoglobin.
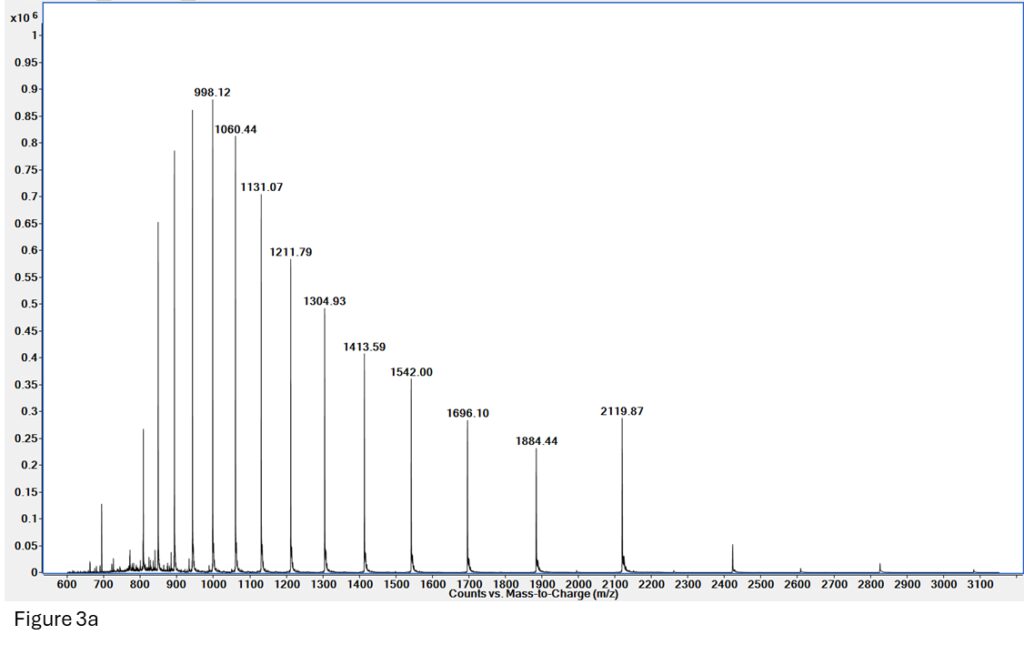
Figure 3a: Spectrum for myoglobin made on a reversed-phase (denaturing) LCMS run. The signal is very clean and gives a great deconvoluted mass for Apomyoglobin (Figure 3b).
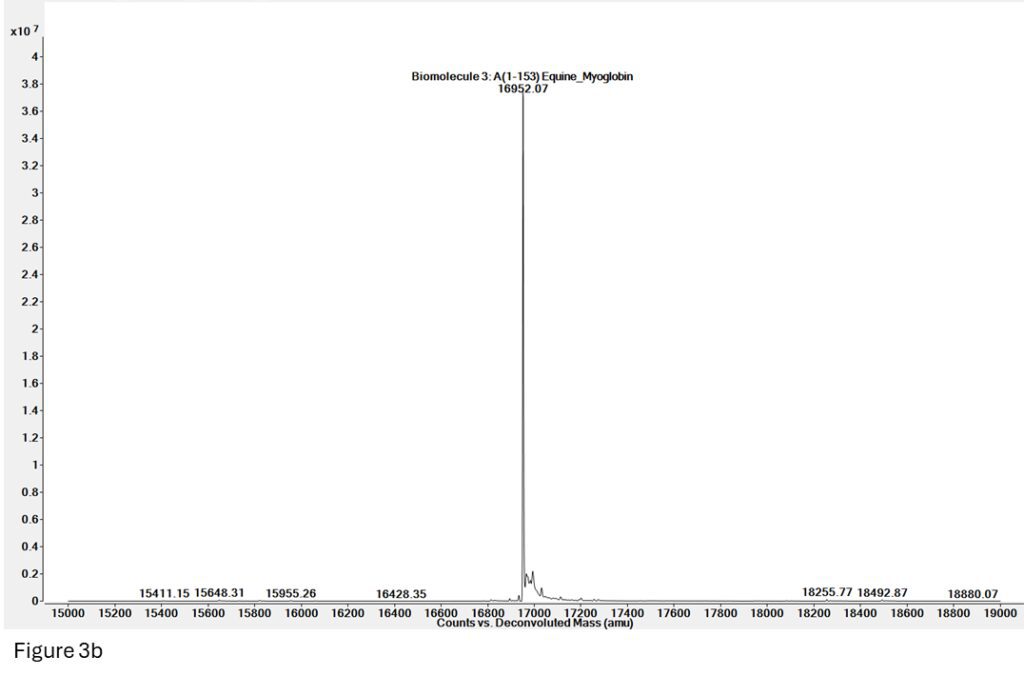
Figure 3b: Deconvoluted mass for Apomyoglobin from spectrum 3a (denatured, mass = 16952), but there is no presence of Holomyoglobin (native, 16952 + 615 = 17567).
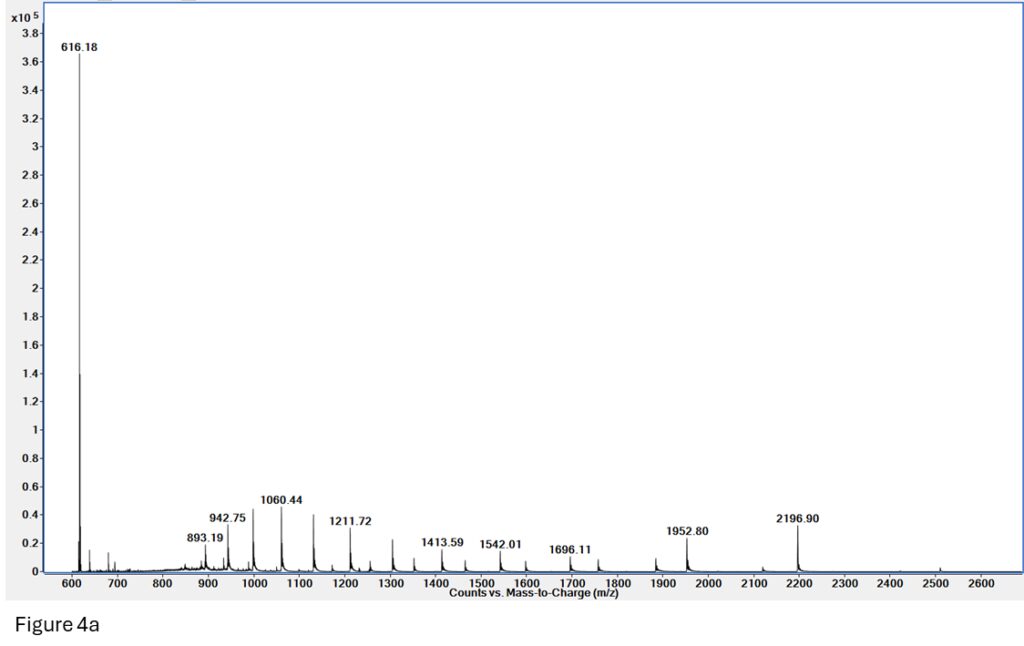
Figure 4a: Native MS on myoglobin. We can see two charge envelopes present – denatured (893 – 1696 m/z) and native (1150 – 2500 m/z), as well as free heme (m/z = 616).
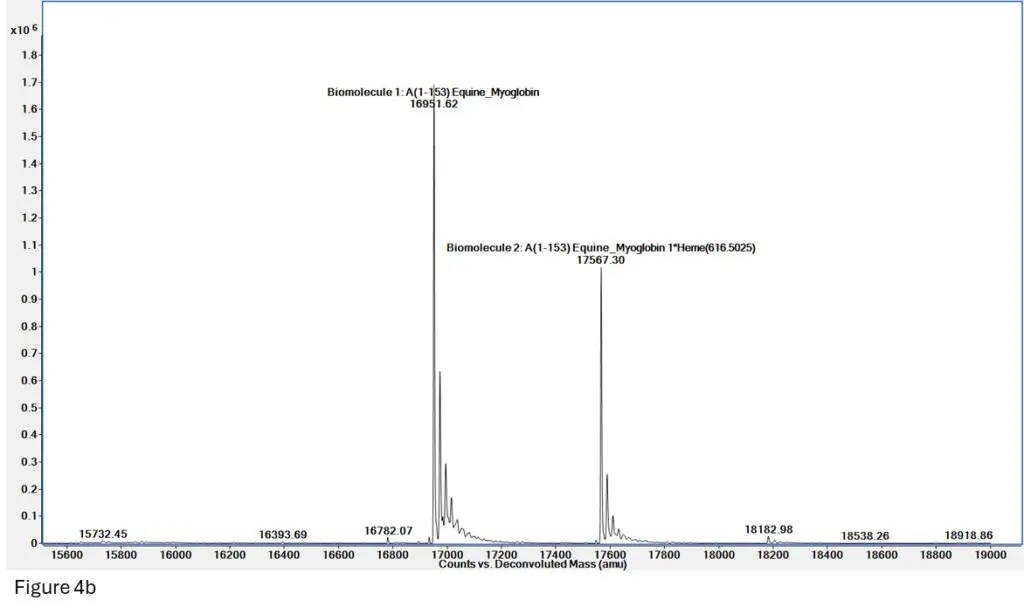
Figure 4b: Deconvolution of the spectrum in Figure 4a shows two very clean masses for both Apomyoglobin (denatured, mass = 16952) and Holomyoglobin (native, 16952 + 615 = 17567), indicating we have successfully maintained the heme-myoglobin interaction through ionization and mass spec
readout.
Conclusion
In the realm of proteomics, intact protein mass spectrometry and native protein mass spectrometry represent two powerful tools for deciphering the intricacies of protein structure, function, and nteractions. Understanding the differences between these techniques allows researchers to choose the most appropriate approach based on their specific research questions and analytical objectives. Together, these methodologies continue to push the boundaries of proteomic research, unveiling the mysteries of the proteome with unprecedented detail and clarity.
{kind=link}